

Pulmonary tuberculosis (PTB) accounts for roughly 85% of the global disease burden caused by Mycobacterium tuberculosis (Mtb), and understanding its mechanisms is vital for improving prevention and treatment outcomes. In a recent study using a mouse model, researchers sought to unravel how lung-specific environments can promote Mtb survival and replication, highlighting the complex interactions between dysplastic macrophages and lung tissue.
Despite the presence of a robust systemic immune response, certain immunocompetent individuals fail to contain Mtb, leading to active pulmonary disease characterized by cavitary lesions from which infectious aerosols can be transmitted. This study focused on the extended repercussions of primary lung lesions, particularly in B6.Sst1S mice — a strain susceptible to tuberculosis due to genetic factors — following hematogenous spread from primary lesions.
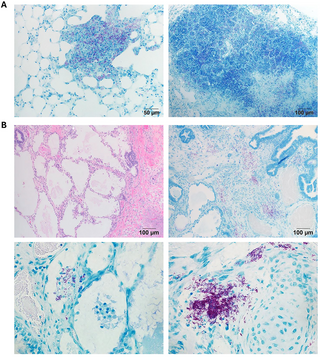
Initially, secondary lung lesions appear to exhibit localized immune responses, with organized tertiary lymphoid follicles akin to those in resistant B6 mice. These early lesions predominantly house non-replicating bacilli, indicating a temporary containment of the bacteria. However, as the disease progresses, notable changes occur, including an increase in myeloid cell populations, differentiation of macrophages, dissolution of lymphoid structures, and the emergence of dysplastic lung epithelial cells. This cascade of events culminates in the development of necrosuppurative pneumonia, mirroring the advanced stages of pulmonary tuberculosis observed in human patients.
The research team conducted fluorescence multiplex immunohistochemistry and spatial transcriptomic analyses on the divergent TB lesions that develop in these susceptible mice. The findings revealed that the inflammation-injured lung and aberrantly activated macrophages actively contribute to the detrimental local microenvironment conducive to the virulent Mtb.
A critical component of this understanding centers around the implications of lung tissue and its cellular composition. When fragments of lung and spleen tissues were implanted subcutaneously prior to infection, the lung implants retained the ability to form organized granulomas with necrosis and Mtb replication, thus showing that the lung’s cellular makeup — rather than oxygen levels — dictates PTB progression.
Additionally, the results indicate that significant interactions between activated macrophages and compromised lung resident cells contribute to the susceptibility of immunocompetent hosts to TB. This insight suggests that these interactions — which appear to be evolutionarily conserved — could serve as potential targets for host-directed therapies aimed at tuberculosis.
The investigation also noted that approximately 5-10% of individuals infected with Mtb would develop active TB later in life, particularly following primary infections acquired during childhood. These findings underscore a need for further research to understand why specific immune responses fail in lung environments and how this variation impacts TB transmission.
The importance of the mice model used is noteworthy, as it enables scientists to mimic the human disease without the need to directly infect the lungs through aerosolized bacteria, which is the usual method in experimental studies.
In conclusion, the study suggests a nuanced understanding of pulmonary TB progression, demonstrating how dysregulated macrophage activation and the maladaptive repair process in lung tissue can create an environment conducive to Mtb replication. Such findings hold promise for developing more effective therapeutic strategies aimed at mitigating tuberculosis pathogenesis and spread.
The conclusions from this study were detailed in the paper ‘Dysplastic lung repair fosters a tuberculosis-promoting microenvironment through maladaptive macrophage polarization,’ published in PLoS Pathogens by Yabaji et al. This work received funding from the National Institutes of Health and emphasizes the collaborative necessity for targeted interventions in the fight against TB.



